Introduction to the Use of Genomic Data in Phylogenetic Systematics
Aula 1
Objetivos
Quando lidamos com dados genômicos, temos que manipular e organizar muitos arquivos de input e output, o que parece muito complicado. Porém, quando sabemos utilizar linha de comando, essas tarefas tornam-se mais simples. O objetivo deste tutorial (e da aula como um todo) é dar uma visão geral sobre como comunicar com seu computador através da linha de comando para executar tarefas que seriam muito complicadas de executar com cliques do mouse. Esperamos que no final da disciplina você tenha uma base para trabalhar com dados de sequenciamento massivo, e para desenvolver melhor suas habilidades em bioinformática, caso tenha interesse no futuro. Lembre-se que o google tem muitas respostas para suas perguntas sobre programação. Só é necessário saber perguntar.
O que é a Shell
Shell é um programa que permite você se comunicar com o sistema operacional do seu computador através de comandos usando o teclado, sem a necessidade de uma interface gráfica (Graphical User Interface, GUI) que você opera com o mouse.
A comunicação com o computador através de GUI é intuitiva e prática para tarefas do dia a dia. Porém, quando há a necessidade de fazer tarefas repetitivas, o shell pode te poupar muitas horas.
Existem vários programas que você pode usar para acessar o shell do seu computador. No Windows, os mais comuns são o Command Prompt e o Power Shell. Em sistemas Unix (Mac, Linux) temos o Bash. A linguagem utilizada pelo shell do Windows é diferente da linguagem utilizada no Unix. Como a maioria dos programas são feitos para Mac e Linux, aqui vamos focar na linguagem Bash.
Se você tem uma máquina com o Windows instalado, não se preocupe. Foi lançado recentemente um aplicativo que permite você abrir um terminal em Bash: o Windows Subsystem for Linux (WSL).
Siga as instruções para instalar e escolha qual distribuiçã do Ubuntu você deseja instalar.
Utilizando a Shell
Abra o WSL (ou o aplicativo chamado Terminal no MAC/Linux).
Você verá algo parecido com a Figura 1.
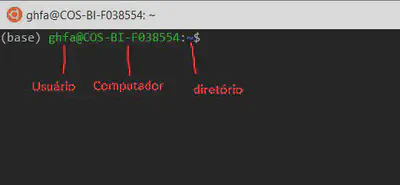
O sinal “~” indica que você está no user home directory. Seria análogo à sua “área de trabalho” no Windows.
Para exibir o caminho completo do diretório em que estamos trabalhando, utilizamos o comando ‘pwd’ (Neste Tutorial, a caixa escura contem os comandos que você deve executar em seu terminal. Recomendo você digitar o comando para memorizar, ao invés de apenas copiar e colar. Linhas com ‘#’ são comentários, não comandos. Elas são ignoradas pela shell.)
pwd
Observe que você estará na pasta “/home/seu_nome_de_usuario”, onde seu_nome_de_usuario é o nome que você escolheu quando instalou o Ubuntu. O “~” é uma simplificação para o home do usuário.
Para navegar pelas pastas, utilizamos o comando ‘cd’ (de change directory). Você pode ir para a pasta home (note que aqui é o home do sistema) digitando:
cd /home
Para saber o que tem nesta pasta em que estamos agora, podemos utilizar o comando ‘ls’ (de list)
ls
Use o comando ‘cd’ para acessar a sua pasta novamente. Você pode usar o nome dela como argumento para o ‘cd’ ou simplesmente usar:
cd ~
Observe que se você usar ‘ls’ nada acontecerá, pois, sua pasta está vazia.
Para criar um arquivo você pode usar o comando ‘touch’.
touch exemplo.txt
Use o comando ‘ls’ agora e veja que o arquivo estará na sua pasta. Existem vários editores de texto. Nano e Vim são bastante comuns e normalmente já vem instalados nos sistemas Unix. Eu particularmente uso o Vim. Uma maneira fácil de saber se um programa está instalado no seu computador é usando o comando ‘which’, seguido pelo nome do programa.
which vim
Caso esteja instalado, o comando retorna o caminho para a o programa, neste caso ‘/usr/bin/vim’
Você pode acessar o manual de um programa com o comando ‘man’ (de manual).
man nano
man vim
Utilize o vim para abrir o arquivo que criamos anteriormente
vi exemplo.txt
Observe que o arquivo está em branco. Para editar você tem que digitar primeiro ‘i’ para ativar o insert mode.
Escreva “Isso é um exemplo de arquivo”
Quando terminar, aperte a tecla “Esc” para sair do Insert. Para salvar digite ‘:wq’ (write and quit). Caso vc queira sair sem salvar alteraçOes no documento, digite apenas ‘:q’.
Leia o manual com calma em algum momento, caso queira saber mais detalhes sobre como usar a aplicação. Tente editar o arquivo com o Nano e veja qual você prefere.
Outra forma de criar um arquivo é através do próprio programa de edição, utilizando como input o nome de um arquivo que não existe.
vi exemplo2.txt
Escreva algo no arquivo, salve e feche-o.
Existem algumas maneiras de ver o arquivo sem editar. Uma delas é utilizando o comando ‘cat’ (de concatenated). Veremos outras formas em breve.
cat exemplo2.txt
Uma forma de escrever em arquivos é utilizando o comando ‘echo’. Este comando mostra um texto no terminal.
echo Olá, tudo bem?
Podemos utilizar o sinal ‘>’ para que o resultado de um comando seja enviado para um arquivo
echo Olá, tudo bem? > exemplo3.txt
vi exmplo3.txt
Caso o arquivo especificado como output não exista, ele será criado. Caso contrário, o que está nele será apagado e substituído pelo novo output.
echo Tudo bem, e com você? > exemplo3.txt
vi exmplo3.txt
Para adicionar algo a um arquivo sem apagar, use ‘»’
echo Olá, tudo bem? >> exemplo4.txt
echo Tudo bem, e você? >> exemplo4.txt
cat exmplo4.txt
Você pode usar o ‘cat’ para concatenar dois arquivos
echo Eu estou bem tambem >> exemplo5.txt
cat exemplo4.txt exemplo5.txt
# para salvar oresultado em um outro arquivo
cat exemplo4.txt exemplo5.txt > exemplo6.txt
cat exemplo6.txt
Use o ‘ls’ para listar os arquivos na pasta
Muitos comandos possuem opções que podem ser utilizadas para controlar melhor o output. Para ver rapidamente como usar um programa/comando sem precisar abrir o manual, podemos utilizar o argumento ‘–help’ ou muitas vezes somente ‘-h’ basta.
ls --help
Para exibir os arquivos em uma lista detalhada, utilize ‘ls -l’
ls -l
A primeira coluna indica o tipo de arquivo e as permissões. Veja a Figura 2 obtida do site Data Carpentry
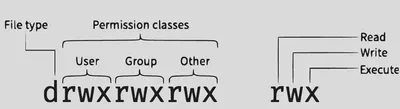
Para mudar as permissões, usamos o comando ‘chmod’
# Para tornar o arquivo executável podemos utilizar o comando abaixo.
# (por exemplo no caso de programa que criou ou baixou)
chmod +x exemplo6.txt
ls -l
Para criar uma pasta usamos o comando ‘mkdir’ (make directory)
mkdir aula_01
mkdir aula_01/pasta1
mkdir aula_01/pasta2
O comando acima cria uma pasta dentro da pasta que havia sido criada anteriormente. Podemos usar o ls seguido de -lR para listar em detalhe e recursivamente o conteúdo dentro as pastas que criamos.
ls -lR
Para copiar arquivos usamos o comando ‘cp’ (copy), seguido do arquivo que queremos copiar e o local de destino
cp exemplo6.txt aula_01/exemplo6.txt
cp exemplo5.txt aula_01/pasta1/exemplo6.txt
ls -lR
Para remover utilizamos o comando ‘rm’ (remove) seguido pelo arquivo que desejamos remover.
rm exemplo6.txt
# Obs: Para remover uma pasta, usamos a opção -r (recusrsively)
rm -r aula_01/pasta1
ls -lR
Lembre-se: o shell não te pergunta se você tem certeza da ação que você está tomando. E também não existe “lixeira”. Uma vez deletado, já era… Tome cuidado e tenha sempre backup de arquivos importantes.
Para mover um arquivo usamos ‘mv’ (move) seguido pelo caminho pro arquivo de origem e pelo destino.
mv exemplo5.txt aula_01/exemplo5.txt
ls -lR
Podemos renomear o arquivo mudando o nome de destino.
mv exemplo4.txt novo_nome_para_o_exemplo4.txt
ls -lh
Podemos usar caracteres coringas (wild cards) para executar operações em muitos arquivos. Por exemplo, para copiar todos os arquivos .txt para uma pasta
cp *.txt aula_01/
ls -l aula_01
# Note que o ls acima lista apenas o conteúdo da pasta aula_01
Remova todos os arquivos e pastas que você criou até agora neste tutorial e limpe o terminal. (Tenha certeza que vc está na pasta certa)
rm -r *
# O ‘-r’ significa recursivamente (para deletar pastas)
# o ‘*’ é um caractere coringa.
# Como não tem nenhum outro caractere especificado,
# ele vai deletar tudo que está na pasta.
clear
# O comando acima limpa o seu terminal
Se você quiser salvar os comandos que você executou, vc pode usar o comando ‘history’. Este comando exibe no terminal todos os comandos utilizados (mesmo comandos utilizados a muito tempo atás). Você pode usar ‘>’ para salvar essa história em um arquivo. Veja a ajuda do programa para como limpar seu histórico.
Mais a frente no tutorial vamos ver comandos que podem ser úteis para salvar apenas os últimos comandos.
history > terminal_history_2021_.txt
Agora que você já tem uma noção de alguns comandos básicos, vamos instalar programas. Ao longo dos tutorias vamos introduzir novos comandos úteis. Você também pode dar uma olhada com calma em outros sites como o Data Carpentry ou SoftwareCarpentry que contém alguns tutoriais e exercícios.
Instalando programas
Muitos programas hoje em dia apresentam bons manuais online que ensinam o passo a passo de como instalar programas. Aqui, vamos ensinar algumas dicas, mas é sempre importante ler em detalhe a página do programa antes de instalá-lo.
Para facilitar, vamos criar uma pasta que usaremos para baixar programas quando necessário.
mkdir progs
cd progs
Uma forma fácil de instalar programas é usando ferramentas que te ajudam a instalar e administrar pacotes de programas (package manager). Uma dessas ferramentas que vem com o Ubuntu é o programa APT. A desvantagem do APT é que é necessário acesso a raiz do sistema (se você não é o administrador a instalação não é possível). Além disso, muitas vezes é necessário instalar outros programas (dependências) antes de instalar o programa desejado. Para usar os privilégios de administrador, você precisa usar o comando ‘sudo’ andes do comando ‘apt’ e fornecer sua senha de usuário criada durante a instalação do programa. Use os comandos abaixo para instalar o programa R em seu sistema Linux. Para mais detalhes da instalação veja este site
# Primeiro atualize o apt
sudo apt update
# Instale as dependências para o R.
sudo apt install dirmngr gnupg apt-transport-https ca-certificates software-properties-common
# Adicione o repositório CRAN para a lista do seu sistema
sudo apt-key adv --keyserver keyserver.ubuntu.com --recv-keys E298A3A825C0D65DFD57CBB651716619E084DAB9
sudo add-apt-repository 'deb https://cloud.r-project.org/bin/linux/ubuntu focal-cran40/'
# Instale o R
sudo apt install r-base
# Instale o programa para poder compilar pacotes do R
sudo apt install build-essential
# Para abrir o R no terminal, apenas digite R
R
# Agora o seu terminal está falando a linguagem R
# Comandos em bash não serão reconhecidos
pwd
# Para sair do R e voltar barra a linguagem bash, digite
q()
# Não salve o workspace.
Um administrador de pacote muito útil é o Conda. Com ele podemos instalar e desinstalar pacotes facilmente e sem acesso à raiz. Embora seja escrito em Python, ele pode ser usado para instalar programas que foram escritos em várias outras linguagens. O Conda vem em dois principais sabores: Anaconda e Miniconda. Miniconda é uma versão menor e mais básica, e geralmente tem tudo o que precisamos. Por isso vamos utilizá-la.
Para instalar o conda precisamos baixar o instalador da internet. Para isso podemos usar o comando ‘wget’.
Abra a página que contém os instaladores para Miniconda e vá até parte correspondente ao seu sistema operacional. (Lembre-se, se vc está usando o WSL, seu sistema operacional é Linux) Click com o botão direito e copie o endereço. No Terminal, digite ‘wget’ e cole o endereço copiado (Não use ‘Ctrl + V’. Apenas clique com o botão direito do mouse) (Lembre-se também de ‘Ctrl + C’ no Unix shell serve para cancelar um processo, não para copiar.)
wget https://repo.anaconda.com/miniconda/Miniconda3-latest-Linux-x86_64.sh
O arquivo baixado é um script em bash que vai instalar o Miniconda. Podemos abri-lo com o Vim ou nano para vê-lo.
vi Miniconda3-latest-Linux-x86_64.sh
Feche o arquivo sem salvar alterações digitando ‘:q’
Execute o arquivo que você baixou. Como é um script em bash, utilizamos:
bash Miniconda3-latest-Linux-x86_64.sh
Siga as intruções e aceite o default.
Agora você pode usar conda para instalar programas. Você pode procurar por pacotes de interesse no repositório do Anaconda ou utilizar a busca no próprio terminal.
Por exemplo, o programa gdown é interessante para baixar arquivos grandes do google drive. O Comando abaixo busca pelo programa
conda search gdown
Para instalar use:
conda install gdown
# Caso queira uma versão específica
conda install gdown=4.2.0
# Caso queira um canal específico para baixar use a opção -c
conda install -c conda-forge gdown
# Para mais detalhes
conda install --help
Uma vantagem do conda é que podemos criar ambientes com pacotes específicos. Por exemplo, se vamos começar um projeto novo e queremos usar sempre determinadas versões de programas, podemos criar um ambiente com esses programas e ativá-lo sempre que formos trabalhar naquele projeto.
Por exemplo, podemos criar um ambiente para o phyluce, programa que utilizaremos para UCEs. (A versão mais recente do Phyluce não está disponível em um repositório conda. Assim, temos que baixar o arquivo).
# Baixe o arquivo para o seu sistema operacional.
# No caso do exemplo abaixo, Linux
# Para MacOS veja:
# https://github.com/faircloth-lab/phyluce/releases
wget \
https://raw.githubusercontent.com/faircloth-lab/phyluce/v1.7.1/distrib/phyluce-1.7.1-py36-Linux-conda.yml
# Use conda para instalar usando arquivo baixado
conda env create -n phyluce-1.7.1 --file phyluce-1.7.1-py36-Linux-conda.yml
Os argumentos ‘env create’ indica que é para criar um ambiente (create environment) O argumento após a opção ‘-n’ determina o nome do ambiente (no caso phyluce-1.7.1, mas poderia ser qualquer um a sua escolha) e a opção ‘–file’ determina que é para usar o arquivo indicado no argumento seguinte (ou seja, não é para o conda buscar na internet e baixar)
Para ativar o ambiente criado use
source activate phyluce-1.7.1
# O programa illumiprocessor faz parte do phyluce
# veja a ajuda desse programam
illumiprocessor –-help
# Para desativar o ambiente
conda deactivate
# Note que agora não é possível usar o illumiprocessor
illumiprocessor –-help
Um outro programa muito usado para instalar pacotes em Python é o Pip. Visite o website e instale o programa.
Ao longo da disciplina instalaremos mais programas. Caso tenha tempo, você pode navegar pelos tutoriais e instalar os programas antecipadamente.
Acessando pastas do Windows com WSL
Embora estejamos utilizando o Linux, podemos querer acessar documentos que estão em pastas do Windows. Os arquivos do Windows ficam “escondidos” na raiz do WSL. Não utilize o WSL para manipular arquivos de sistema do Windows, use apenas para arquivos comuns do seu projeto.
Lembre-se que estamos na pasta “progs”. Podemos conferir usando ‘pwd’. Para voltar um nível na pasta podemos utilizar o atalho ‘..’
cd ..
Temos que chegar na raiz do sistema. Podemos utilizar várias vezes o ‘cd ..’ ou podemos utilizar o atalho ‘/’ (que sempre te leva pra raiz)
cd /
# visualize o conteúdo do diretório
ls -l
A unidade de disco estará na pasta ‘mnt’
cd mnt
Acesse a unidade de disco em que o Windows está instalado (Possivelmente unidade ‘c’) ou a unidade em que prefira trabalhar com seus arquivos (por exemplo se você tem uma unidade maior para manter arquivos e outra apenas para o sistema operacional). Neste tutorial vamos acessar a unidade em que o Windows está instalado para usar a área de trabalho do Windows, mas você pode escolher a pasta que preferir em seu computador.
cd c
ls -l
Procure por uma pasta chamada “Users” ou “Usuários”. Dentro dela terá uma outra pasta com o seu nome de usuário do Windows (Substitua no comando abaixo pelo nome adequado).
cd Users
ls
cd nome_do_seu_usuario_windows
ls
cd Desktop
# Ou simplesmente
cd Users/nome_do_seu_usuario_windows/Desktop
ls
Observe que todos os seus arquivos da área de trabalho do Windows aparecem listados no terminal. Crie uma pasta na área de trabalho (ou no seu local preferido) para a disciplina
mkdir filogenomica
Dicas:
- Não use espaço nem caracteres especiais (‘, ~, !, * …) ’_’ ou ‘-’ estão OK (mas melhor evitar ‘-’)
- Use nomes curtos e fáceis (porém informativo) para pastas e arquivos
- Lembre-se que “filogenomica” é diferente de “Filogenomica”
Para ficar mais fácil acessar essa pasta do WSL, vamos criar um atalho na pasta /home/$USER do Linux.
Primeiro, acesse a pasta da disciplina e imprima no terminal o seu caminho completo.
cd filogenomica
pwd
Copie (selecione e clique com o botão direito) o caminho para a pasta Volte para a pasta home do seu usuário de Linux. Lembre-se que podemos usar o atalho ‘~’
cd ~
Use o comando ‘ln’ (link) para criar um link para a pasta filogenomica. Como estamos copiando uma pasta, o comando exige que usemos links simbólicos com a opção ‘-s’. O argumento utilizado é o caminho completo para a pasta (ou arquivo) que queremos criar o link. Você pode copiar e colar (selecione e clique com o botão direito duas vezes) o caminho para a pasta resultante do ‘pwd’ usado acima.
ln -s /mnt/c/Users/seu_usuario_windows/Desktop/filogenomica
ls -l
Observe que a cor da pasta é diferente e o caminho para a pasta é indicado ao lado. Sempre que quisermos acessar essa pasta do terminal WSL podemos usar:
cd ~/filogenomica
Ou podemos acessar ela pelo Windows normalmente
Na pasta filogenomica use a linha de comando para criar as pastas “data”, “docs”, “scripts” e “misc”
Crie o arquivo “bash_cheatsheat.csv” na pasta “misc”
Da maneira que preferir (mas usando o terminal), escreva nesse arquivo o seguinte (Não use espaço): Comando,descrição,opções_uteis,uso
Use esse arquivo para ter acesso rápido a comandos úteis que você vai utilizar frequentemente. Adicione os comandos que você aprendeu hoje. Ao longo da disciplina você pode usar o comando ‘echo’ com o output ‘» ~/filogenomica/misc/bash_cheatsheat.csv’ para escrever nesse arquivo sempre que quiser.
No Windows, você pode abrir o arquivo usando um editor de texto (sugiro o notepad++) ou até mesmo o Excel ou outro editor de planilhas.
Obtendo Sequências – Sequence Read Archive
Sequence Read Archive (SRA) é um repositório do GenBank para sequencias cruas resultantes de sequenciamento massivo (high throughput sequencing) de bibliotecas genômicas. Você pode navegar pelo site do SRA para buscar sequencias dos organismos que você tem interesse a baixar pelo site. Porém, uma vez que temos o número de acesso, é mais fácil utilizar uma ferramenta chamada SRA Toolkit. Podemos instalar usando ‘wget’ para baixar o arquivo, como mostrado nas instruções do GitHub do programa, ou podemos usar o apt-get ou o conda. A versão no repositório do conda não é a mais recente:
conda search sra-tools
# Compare o resultado do comando acima com a versão indicada no site:
# https://github.com/ncbi/sra-tools/wiki/01.-Downloading-SRA-Toolkit
Par facilitar, usaremos o conda. Caso queire a mais recente, siga as instruções no site do programa
conda install sra-tools
# Caso queira instalar no mesmo ambiente em que o Phyluce está instalado,
# ative o ambiente primeiro e instale usando o comando acima.
# Caso queira instalar em um ambiente diferente use:
conda create -n nome_do_ambiente sra-tools
# Se for o caso, lembre-se de ativá-lo antes de usar.
O SRA Toolkit possui uma ferramenta chamada ‘fastq-dump’. É ela que usaremos para baixar as sequencias. As sequências que utilizaremos são de aranhas saltadoras publicadas por Leduc-Robert and Maddison, 2018. Para baixar, precisamos do número de acesso da corrida que pode ser encontrado na página (Figura 1).
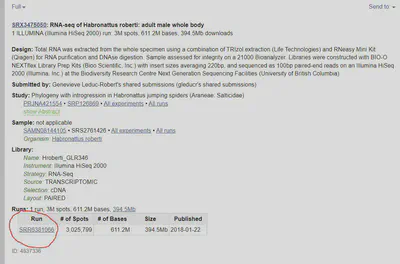
Acesse a pasta “data” que você criou no tutorial passado e crie uma outra pasta para os dados de RNA-seq.
cd ~/filogenomica/data
mkdir raw-rnaseq
cd raw-rnaseq
Vamos utilizar as corridas: SRR6381066, SRR6381055, SRR6381090, SRR6381065.
A forma mais simples de baixar os das é apenas usar o comando ‘fastq-dump’ seguido pelas número de acesso. Porém, muitas vezes é melhor já preparar um pouco o arquivo para usar como input para outros programas. Podemos fazer isso da seguinte maneira:
fastq-dump --defline-seq '@$ac $ri $rl' --defline-qual '+$ac $ri $rl' --gzip --clip \
--split-files --qual-filter -v \
SRR6381066 SRR6381055 SRR6381090 SRR6381065
Onde:
–defline-seq ‘@$ac $ri $rl’: renomeia cada sequencia no arquivo usando variáveis do SRATools (número de acesso, número da leitura, e tamanho da leitura). Alguns programas podem ter problemas com a forma que o fastq-dump nomeia as sequencias, por isso renomeamos. Essa maneira parece funcionar para mim, mas caso tenha problemas, você pode mudar essa opção. Veja o manual e a ajuda do programa para opções.
–defline-qual ‘+$ac $ri $rl’: o mesmo que o anterior, porém renomeia a linha antes a qualidade da sequência.
–gzip: opção para compactar o arquivo.
–clip: opção para remover adaptadores das sequências
–split-files: separa as leituras em dois arquivos diferentes, uma para a primeira leitura, outro para a segunda leitura
–qual-filter: remover leituras e partes das sequencias com qualidade ruim.
-v: verbose, serve para o programa relatar informações no terminal enquanto processa.
SRR6381066 SRR6381055 …: lista de números de acesso.
No tutorial seguinte vamos processar esses dados que baixamos
Aula 2
Objetivos
Introduzir ao processamento de dados gnômicos obtidos com sequenciamento massivo usando como exemplo bibliotecas de transriptoma (RNA-seq. Embora RNA-seq tenha muitas outras utilidades (e.g. estudo de expressão diferencial) vamos focar no processamento dos dados para filogenômica. Ao final do tutorial teremos sequencias de genes ortólogos alinhados.
Arquivo FASTQ
No tutorial anterior, baixamos algumas seuqencias no formato FASTQ. Entre na pasta na qual eles foram salvos para que poçamos examiná-los.
cd ~/filogenomica/data/raw-rnaseq
Como o arquivo está compactado (.gz) os editores que vimos podem não funcionar. Podemos usar o ‘gzip’ para descompactar um arquivo. Porém, como trabalhamos com arquivos muito grandes, melhor mantê-los compactados. Quando temos arquivos muito grandes e compactados (.gz) o programa ‘zless’ é eficiente para visualização. Veja um dos arquivos que acabamos de baixar:
zless SRR6381066_1.fastq.gz
Note que, na primeira e terceira linha de cada sequência, temos as informações que pedimos para serem alteradas com as opções –defline-seq –defline-qual.
Uma outra forma de visualizar os arquivos é com os comandos ‘head’ e ‘tail’, que imprimem no terminal as primeiras ou últimas linhas dos arquivos. Novamente, como o formato é .gz, esses comandos não vão funcionar diretamente. Porém, podemos usar um pipe para descompactar temporariamente o arquivo e executar o comando.
gunzip -c SRR6381066_1.fastq.gz | head -n 16
Onde ‘-c’ indica para o programa gunzip que não é para mudar o arquivo original e apenas descompactá-lo e manter o resultado provisoriamente em um standard output na memória. O sinal ‘|’ (pipe) significa que o output do programa anterior vai ser utilizado como input para o seguinte comando, ‘head’. A opção ‘-n’ indica que é para imprimir apenas as 16 primeiras linhas (o padrão do comando é 10).
O comando ‘tail’ funciona da mesma maneira, porém imprimindo as últimas linhas do arquivo.
Use o que você aprendeu até agora para criar um arquivo com as 80 últimas linhas do arquivo SRR6381066_1.fastq.gz. Salve esse arquivo com “ultimas_linhas.fastq” na pasta “exercicios” dentro da pasta filogenomica.
Essa mesma ideia pode ser usada com o comando ‘cat’ (que imprime todas as linhas) para contarmos o número de sequências em cada arquivo. Lembre-se que cada sequencia em um arquivo fastq é representado por 4 linhas. Podemos usar o comando wc (word count) com a opção ‘-l’ para contar as linhas do arquivo aberto com o ‘cat’.
gunzip -c SRR6381066_1.fastq.gz | cat | wc -l
# O resultado é o número total de linhas.
# Divida por quatro e saberá o número de leituras do arquivo.
Se quisermos fazer isso várias vezes para todos os arquivos na pasta, podemos usar um loop com wild cards e variáveis. Variáveis são criadas com o sinal “=” e são chamadas colocando “$” na frente do nome escolhido para a variável. Caso contenham algum comando ou operação, determinamos a variável com “=$(comando)” Exemplo:
Nome='Guilherme'
Idade=35
Cidade='Belo Horizonte'
Nascimento=$((2021-$Idade))
echo Olá, me chamo $Nome, tenho $Idade anos e nasci em $Cidade no ano de $Nascimento.
Arquivos=$(ls -1 | wc -l)
echo Nesta pasta tem $Arquivos arquivos
O que queremos nesse caso é: para cada arquivo “i” na pasta terminado em .fastq.gz, usar o gunzip em um pipe para imprimir (invisivelmente) todo o arquivo e contar o número de linhas. Depois queremos dividir esse número de linhas por 4 e queremos que o nome do arquivo seja impresso no terminal seguido pelo número de leituras. Traduzindo isso para bash, podemos falar assim:
for i in *.fastq.gz
do
lines=$(gunzip -c $i | cat | wc -l)
seqs=$(($lines/4))
echo $i $seqs
done
Limpeza e preparação das sequencias
Agora que temos as sequencias, vamos remover leituras com qualidade ruim e que contenhas adaptadores. Para isso usaremos o programa trimmomatic. A instalação do Phyluce automaticamente instala o trimmomatic. Você pode simplesmente carregar o ambiente em que o Phyluce está instalado e utilizar os trimmomatic. Caso precise, ou queira, instalar em um ambiente diferente, você pode usar o conda ou a forma que achar melhor.
conda install trimmomatic
Existem vários parâmetros que podem ser explorados no trimmomatic, porém aqui vamos usar as opções similares as default do phyluce. Para maiores detalhes consulte o manual e a página do programa. Um dos parâmetros que se deve prestar atenção é o ILLUMINACLIP, que busca por adaptadores. Ele depende de como a biblioteca foi construída. Aqui vãos usar o padrão “TruSeq3-PE”. Baixe o arquivo fasta da página do GitHub:
cd ~/filogenomica/data
wget https://github.com/usadellab/Trimmomatic/blob/main/adapters/TruSeq3-PE.fa
Esses adaptadores são geralmente usados pelas plataformas HiSeq e MySeq), mas pode não ser adequado para algumas bibliotecas. Caso não saiba exatamente quais adaptadores foram usados, você pode criar arquivos customizados, ou usar um arquivo concatenando todos os tipos de adaptadores disponibilizados pelo trimmomatic (veja o manual.
O trimmomatic processa um arquivo de cada vez. Para ser mais eficiente, vamos fazer um loop para facilitar o nosso trabalho.
Primeiro, vamos criar uma pasta para o output chamada “clean_rnaseq” na pasta filogenomica/data
cd ~/filogenomica/data
mkdir clean_rnaseq
Crie um arquivo csv em que cada linha tenha as leituras e o nome das amostras que você quer processar separado por vírgulas. Salve-o como “inputs.csv”. Você pode fazer no excel ou em um arquivo texto. Pode usar também a linha de comando, caso queira praticar.
echo 'SRR6381066_1.fastq.gz,SRR6381066_2.fastq.gz,Habronattus_roberti
SRR6381055_1.fastq.gz,SRR6381055_2.fastq.gz,Habronattus_sansoni
SRR6381090_1.fastq.gz,SRR6381090_2.fastq.gz,Habronattus_aztecanus
SRR6381065_1.fastq.gz,SRR6381065_2.fastq.gz,Habronattus_altanus
' > arquivo_inputs.csv
Agora, vamos usar esse arquivo para criar uma variável na shell.
Para criar a nossa variável com os inputs para o trimommatic podemos usar:
Inputs=$(cat arquivo_inputs.csv)
echo $Inputs
Precisamos também criar uma variável com caminho para a pasta com os arquivos fastq e para a pasta de output
rawreadspath=~/filogenomica/data/raw-rnaseq
outdir=~/filogenomica/data/clean_rnaseq
Agora podemos iniciar nosso loop. O que queremos fazer é: para cada elemento “i” na lista “Imputs” vamos utilizar o primeiro elemento antes da vírgula como argumento para o arquivo com as leituras 1, o segundo elemnto como o arquivo com as leituras 2, e o terceiro elemento como nome do output. Também queremos que o output esteja organizado de maneir que possa ser utilizado pelo phyluce (vocês verão detalhes mais a frente). Traduzindo isso para a linguagem bash fica assim:
for i in $Inputs
do
# Seleciona o primeiro elemento de $i delimitado pela vírgula e cria
# uma variável para ele chamada r1
r1=$(echo $i | cut -f1 -d",")
# Segundo elemento
r2=$(echo $i | cut -f2 -d",")
# Último element, que é o nome da espécie
sp_name=$(echo $i | cut -f3 -d",")
# Imprime no terminal uma mensagem só para saber qual arquivo o loop está processando
echo "Processing "$sp_name""
# aciona o trimmomatic para processar a amostra $i.
# Ajuste o número de threads para o seu computador
trimmomatic PE -threads 14 \
$rawreadspath/$r1 \
$rawreadspath/$r2 \
$sp_name"-READ1.fastq.gz" \
$sp_name"-READ1-single.fastq.gz" \
$sp_name"-READ2.fastq.gz" \
$sp_name"-READ2-single.fastq.gz" \
ILLUMINACLIP:TruSeq3-PE.fa:2:30:10:2:keepBothReads \
LEADING:5 TRAILING:15 SLIDINGWINDOW:4:15 MINLEN:40
# Como o phyluce junta as sequencias 1 e 2 que não possuem par,
# faremos o mesmo usando o cat e removemos os arquivos originas sem par
cat $sp_name"-READ1-single.fastq.gz" \
$sp_name"-READ2-single.fastq.gz" \
> $sp_name"-READ-singleton.fastq.gz"
rm $sp_name"-READ1-single.fastq.gz" $sp_name"-READ2-single.fastq.gz"
# Organizamos os arquivos em pastas de maneira necessária para o Phyluce
mkdir $outdir/$sp_name
mkdir $outdir/$sp_name/split-adapter-quality-trimmed
cd $outdir/$sp_name/split-adapter-quality-trimmed
mv ../../../*READ*.fastq.gz .
cd ../../..
done
No final você deverá ter uma pasta para cada espécies, e dentro desta pasta, uma pasta chamada “split-adapter-quality-trimmed” com as sequencias limpas.
Aula 3
Controle de Qualidade
No tutorial anterior, fizemos a limpeza das sequencias. Provavelmente muitas foram removidas por baixa qualidade ou tamanho.
Pense em uma maneira de contar o número de sequências em cada arquivo, como feito anteriormente para as raw reads. Compare os números antes e depois do trimmomatic.
Para verificar a qualidade das sequencias, podemos utilizar o FastQC. Use o conda para instalar o programa.
conda install fastqc
O FastQC tem como input uma lista de arquivos fastq. Ele busca por problemas comuns, como a presença de adaptadores e baixa qualidade de sequencias. Como nossos arquivos estão organizados em pastas diferentes, temos que criar um loop para acessar cada pasta e usar os arquivos como input. Também queremos copiar toso os outputs para uma única pasta. Tente pensar em uma forma de fazer isso, antes de olhar a forma como eu faço. Olhe a ajuda do fastqc para saber das opções.
cd ~/filogenomica/data
mkdir fastqc_output
cd fastqc_output
out_dir=$(pwd)
cd ../clean_rnaseq
species_folders=$(ls)
for species in $species_folders
do
cd $species/split-adapter-quality-trimmed
fastqc -o $out_dir *.fastq.gz
cd ../..
done
cd $out_dir
rm -r *.zip
cd ..
Tente entender o que foi feito acima, o que significa cada comando. Abra os resultados (no Windows) e compare os resultados com o que é explicado no manual do programa.
Assembly
Dois programas são mais comumente usados para fazer o assembly: Trinity e o SPAdes. Ambos podem ser instalados facilmente com o conda. Estudos mostram que os dois são muito similares e funcionam bem na maioria dos casos. Dependendo do objetivo, pode ser mais interessante usar um ou outro (e.g. para quantificar isoformas o Trinity pode ser um pouco melhor). Para filogenias em que desejamos ter muitos genes ortólogos de cópia única, o SPAdes frequentemente funciona melhor (Holzer and Marz, 2019). Por experiência própria tenho preferido o SPAdes (geralmente recupera mais loci e os loci são mais longos), mas você pode experimentar e ver o que funciona melhor pros seus dados.
O SPAdes possui vários módulos (spades, rnaspades, metaspades…). Para RNA podemos usar tanto o spades quanto o rnaspades, sendo que o spades com algumas configurações específicas pode ser melhor que o rnaspades (Holzer and Marz, 2019). Aqui vamos usar dessa forma para o assembly das sequencias limpas que processamos anteriormente. O spades é instalado com o phyluce e você pode usá-lo ativando o ambiente do phyluce (mas fique atento a versão, se não me engano ele pode não ser a mais recente). Alternativamente, você pode instalar em outro ambiente. A versão em que este tutorial foi testada é a 3.15.2 e pode não funcionar com versãos antigas (3.13 por exemplo).
conda install spades=3.15.2
O SPAdes tem com input as sequências limpas (read 1, read 2 e single). Vamos fazer um loop para entrar em cada pasta e processar os arquivos, enviando o output para uma outra pasta.
O programa possui muitas opções que podem ser exploradas (veja as instruções no site. Aqui vamos usar a opção sugerida por (Holzer and Marz, 2019) e similar a apresentada no phyluce: –sc –careful –cov-cutoff auto. A opção
- ‘–sc’ (single cel) é adequada para RNAseq e UCEs pois a cobertura do genoma é desigual.
- ‘–careful’ corrige pra remover indels curtos (pode não ser bom para genomas grandes).
- ‘–cov-cutoff auto’ remove contigs com baixa cobertura selecionando um limite automaticamente.
cd ~/filogenomica/data
mkdir spades_assembly
cd spades_assembly
outfolder=$(pwd)
cd ..
cd clean_rnaseq
speciesfolders=$(ls)
# ajuste no commando abaixo a memória e o número de threads (-t) para seu computador
for i in $speciesfolders
do
echo "Processing "$i""
spades.py --sc --careful --cov-cutoff auto --memory 24 -t 12 \
-1 $i/split-adapter-quality-trimmed/*-READ1.fastq.gz \
-2 $i/split-adapter-quality-trimmed/*-READ2.fastq.gz \
-s $i/split-adapter-quality-trimmed/*-READ-singleton.fastq.gz \
-o $outfolder/$i
done
Obs: Para algumas sequencias do conjunto de dados escolhidos, o arquivo com single reads pode ter 0 sequencias. Nesse caso, remova a linha -s do comando do SPAdes
O Assembly pode demorar um pouco dependendo do computador e do tamanho de cada arquivo.
Aula 4
Assembly - continuação
No último tutorial deixamos o SPAdes processando. Caso não tenha terminado, cancele o processo e baixe a pasta com os resultados para seguir.
Certifique-se que está na pasta que escolhemos para output
cd ~/filogenomica/data/spades_assembly
Observe que o output do SPAdes gera muitos arquivos e pastas para cada espécie.
ls -lh
ls -lh Habronattus_roberti
# note a opção -lh (human readable) para mostrar o tamanho dos arquivos em KBytes.
Verifique o output para ver se ouve algum problema. Podemos olhar as últimas linhas do arquivo spades.log. Caso algo de diferente tenha ocorrido durante o processo, o programa salva um arquivo chamado “warnings.txt”. Você pode verificar em todas as pastas qual espécie teve warning com um loop e usando o ‘grep’. Podemos usar o comando ‘grep’ para imprimir linhas que tenha um determinado caractere ou palavra. Veja a ajuda do comando e observe a opção ‘-c’. Com ela ao invés de imprimir a linha, podemos contar quantas vezes a linha com o padrão aparece. Usamos o ’ls’ para listar os arquivos e o grep para contar quantas vezes o arquivo “warnings.log” aparece em cada pasta.
for i in $(ls)
do
warning=$(ls $i | grep -c "warnings.log")
echo $i $warning
done
O comando acima imprimiu o nome das espécies com 0 para as que não tiveram warning e 1 para quem teve. Verifique a mensagem.
O arquivo que realmente nos interessa é o “contigs.fasta”. Queremos copiar todos esses arquivos para uma única pasta e renomeá-los com o nome de cada espécie.
Primeiro, vamos criar uma pasta para os contigs.
cd ~/filogenomica/data
finalcontigsfolder=spades_contigs
mkdir $finalcontigsfolder
Agora criamos uma variável do tipo ARRAY (uma lista, ou um vetor) com os nomes das pastas na pasta de output do SPAdes. Se você está fazendo de forma seguida e não fechou o terminal, no loop anterior, você já criou uma variável com o caminho para a pasta de output (“$outfolder”) você pode usar essa mesma variável par o loop. Caso contrário, defina a variável novamente.
cd ~/filogenomica/data
outfolder=spades_assembly
resultsfolder=$(ls $outfolder)
for i in $resultsfolder
do
cp $outfolder/$i/contigs.fasta $finalcontigsfolder/$i.fasta
done
Pronto! Agora temos os contigs em uma só pasta organizado em um arquivo fasta por espécie. Podemos contar o número de contigs por espécie. Em um arquivo fasta, as seuquencias são determinadas pelo sinal “>”. Podemos usar o comando ‘grep’ para imprimir linhas que tenha um determinado caractere ou palavra. Veja a ajuda do comando e observe a opção ‘-c’. Com ela ao invés de imprimir a linha, podemos contar quantas vezes a linha com o padrão aparece. Você pode usar um caractere coringa para contar em todas as espécies na pasta
grep -c ">" spades_contigs/*.fasta
Você pode usar seus conhecimentos para salvar esse resultado em um arquivo .txt.
Tente fazer o mesmo, porém usando a opção RNA do SPAdes. Use a ajuda do programa (‘spades.py –help’) e o manual.
Tente também instalar e utilizar o Trinity de maneira similar à que usamos aqui! Como o output do Trinity é um pouco diferente, você terá que fazer mais ajustes em seu script. Compare os resultados dos três métodos.
Removendo Isoformas
Um gene pode ser transcrito de várias maneiras através de splicing alternativo. Quando usamos o Trinity e o rnaSPAdes, possíveis isoformas são identificadas no processo. Para filogenias, isoformas não nos interessa. Embora dependendo do método que usamos possa não atrapalhar na detecção de ortologia, a redundância de sequencias pode tornar o processo mais demorado. O Trinity possui um script auxiliar retém somente a isoforma mais longa. Você pode baixar o script aqui e usá-lo caso tenha feito o assembly com o Trinity ou com rnaSPAdes. Adapte o loop abaixo para o seu caso. Caso não tenha rodado o Trinity e o rnaSPAdes, pule o script abaixo.
cd ~/filogenomica/data
wget \
https://github.com/trinityrnaseq/trinityrnaseq/blob/master\
/util/misc/get_longest_isoform_seq_per_trinity_gene.pl
# Crie uma pasta para output e uma variável para o caminho
mkdir contigs_isormoved
cd contigs_isormoved
output_folder=$(pwd)
cd ..
#defina uma variável o caminho para apasta
folder_with_contigs=caminho/para/a/sua/pasta/com/os/contigs
filelist=$(ls $folder_with_contigs)
cd $folder_with_contigs
for file in $filelist
do
perl ../get_longest_isoform_seq_per_trinity_gene.pl $file > $output_folder/$file
done
Como usamos o SPAdes no exemplo, o script acima não vai funcionar. Para remover as redundâncias de assemblies feito com SPAdes, vamos utilizar o CD-HIT.
cd ~/filogenomica/data
conda install cd-hit
# Defina a pasta de input como variável
cd spades_contigs
inputfolder=$(pwd)
cd ..
# defina a paste de output
mkdir spades_contigs_cd-hitout
cd spades_contigs_cd-hitout
output=$(pwd)
cd ..
names=$(ls $inputfolder)
for file in $names
do
cd-hit-est -i $inputfolder/$file -o $output/$file \
-c 0.95 -n 10 -M 0 -T 20 -sf 1
done
# O programa gera dois arquivos de output para cada arquivo de input
# Para facilitar, vamos mover os arquivos .clstr para outra pasta
mkdir spades_contigs_cd-hitout_clstrfiles
mv spades_contigs_cd-hitout/*.clstr spades_contigs_cd-hitout_clstrfiles
Quantas sequencias foram removidas? Use seus conhecimentos para criar um arquivo com o número de sequencias para cada espécie antes e depois.
Obs: O CD-HIT, embora seja bastante usado, pode não ter uma boa performance Chen et al., 2019, [Behera et al., 2020] (https://doi.org/10.1145/3388440.3412424). Quando o genoma possui muito introns longos ele pode ser difícil fazer alinhamentos como explicado no manual. Entretanto, a maioria dos testes é voltada pra expressão diferencial, e poucos são voltados pra análises filogenéticas. O SPAdes com opção ‘–sc’ recupera bem ortólogos de cópia única, e tende a agrupar isoformas em um mesmo contig (Holzer and Marz, 2019). Por tanto, a combinação SPAdes-sc com CD-HIT pode ser eficiente.
Aula 5
Inferindo Ortologia
Existem vários métodos para inferir ortologia dos contigs revisados em Fernandez et al. (2020) e Nichio et al. (2017). Filogenias geradas com transcriptoma podem ser sensíveis a inferência de ortologia em alguns casos Cheon et al., 2020. Aqui vamos usar dois métodos comumente usados e que mostram eficientes:OrthoFinder e BUSCO.
OrthoFinder
Instale o OrthoFinder usando conda
conda install orthofinder
Orthofinder é muito simples de executar. Só precisamos de um script complementar para adicionar o nome da espécie no cabeçalho das sequencias do arquivo de contigs. Para isso vamos usar o comando ‘sed’ (Stream Editor). A ajuda e o manual não são muto claros de como usar mas tem boas explicações na internet como esta ou esta. Uma forma simples de usar o comando é ‘sed -i “s/o_que_sai/o_que_entra/g” arquivo.alvo’ onde:
-i indica que é para fazer a substituição no arquivo original (ao contrário de imprimir na tela)
s indica que a operação é de substituição
/ indica o limite entre as expressões que serão substituídas
g indica que é para realizar a operação em todos as ocorrências da linha (global)
Exemplo:
echo Olá, eu sou o Guilherme. > arquivo_exemplo.txt
sed "s/Guilherme/Doug/" arquivo_exemplo.txt
# Note que o arquivo não foi modificado, embora na tela tenha sido mostrado a substituição
cat arquivo_exemplo.txt
# Para substituir no arquivo usamos o -i
sed -i "s/Guilherme/Doug/" arquivo_exemplo.txt
cat arquivo_exemplo.txt
# Se não usamos o g temos:
sed -i "s/ /_/" arquivo_exemplo.txt
cat arquivo_exemplo.txt
# Usando o g
sed -i "s/ /_/g" arquivo_exemplo.txt
cat arquivo_exemplo.txt
rm arquivo_exemplo.txt
Abra o arquivo do assembly e veja que as sequencias tem nomes como “>NODE_1_length_6859_cov_1401.737360” e queremos que fique como “>Genero_especie | NODE_1_length_6859_cov_1401.737360”. Antes de olhar a maneira sugerida abaixo, pense como você faria.
O que sugerimos aqui é simplesmente substituir o “>” por “>Genero_especie | “.
Primeiro, vamos criar uma pasta para o output, copiando a pasta original com outro nome, para garantir que caso algo dê errado, não percamos o que fizemos no último passo.
cd ~/filogenomica/data
cp -r spades_contigs_cd-hitout spades_contigs_cd-hitout_renamed
cd spades_contigs_cd-hitout_renamed
files=$(ls)
for file in $files
do
echo "Processing $file"
name=$(echo $file | cut -f1 -d".")
string_to_add=">"$name" | "
sed -i "s/>/$string_to_add/g" $file
done
cd ..
Veja o resultado usando o comando que achar melhor.
Orthofinder tem muitas opções que você pode entender melhor no site. Aqui usaremos da maneira mais simples, que toma como input a pasta com as sequencias. Também temos que especificar que é DNA e não aminoácido com a opção ‘-d’, e o número de processadores (-t). Podemos também determinar o output com ‘-o’ embora não seja necessário
cd ~/filogenomica/data
orthofinder -d -f spades_contigs_cd-hitout_renamed -t 14 -o OrthoFinderResults
Leia na página do programa detalhes sobre o output. Para análises filogenéticas em geral estamos mais interessados nos genes de cópia única que estarão na pasta * Single_Copy_Orthologue_Sequences*. Nela temos as sequencias prontas para serem alinhadas.
Obs: O OrthoFinder busca por cópias únicas que estão presentes em todas (ou quase todas) as espécies (100% completeness). Em alguns casos, podemos ter como resultado 0 ortólogos de cópia única, mas pode existir muitos deles se aceitarmos mais espécies faltantes em cada ortogrupo. De qualquer forma, pode ser interessante permitir genes com espécies faltantes para aumentar o número de genes. Nesse caso é necessário scripts um pouco mais complicados que não vou abordar nessa aula. Caso tenha poucas espécies e genes, você pode abrir o output Orthogroups.GeneCount.tsv na pasta Orthogroups para filtrar os genes e copiá-los da pasta Orthogroup_Sequences para uma nova pasta.
BUSCO
Instale o BUSCO usando conda
conda install orthofinder
Para mais detalhes sobre as opções do BUSCO, verifique o manual. Aqui usaremos a mais simples no modo transcriptoma (‘-m transcriptome’). O BUSCO utiliza um banco de dados de genes curados baseados em alguns genomas. Devemos escolher o banco de dados que contenha os organismos mais próximos do seu grupo. Você pode ver a lista de datasets com o comando:
busco --list-datasets
Se você está usando o dataset de aranhas que baixamos, deverá escolher arachnida_odb10. Lembre-se de ajustar o número de cpus (-c).
cd ~/filogenomica/data
busco -i spades_contigs_cd-hitout -o BuscoResults -m transcriptome -l arachnida_odb10 -c 14
Na pasta de output, veja que há uma pasta para cada espécie. Os genes de cópia única estão na pasta “BuscoResults/Genero_specie/run_arachnida_odb10/busco_sequences/single_copy_busco_sequences”. Observe que temos um arquivo por gene e em cada arquivo temos uma sequência (pois são de cópia única). Como um mesmo banco de dados foi usado para todas as espécies, os nomes dos genes são os mesmos, e, portanto, o nome do arquivo é o mesmo. Note que utilizamos a pasta de contigs sem o nome das espécies, pois o BUSCO não aceita forma como nomeamos. Por isso, temos renomear o output antes de juntarmos todas as espécies em um arquivo por gene. Olhe os outputs e pense em como você poderia fazer isso. Tente fazer, caso não consiga, abaixo segue um script. Tente entender o que o script está fazendo.
cd BuscoResults
folder_list=$(ls -d *.fasta)
n_species=$(ls -1 -d *.fasta | wc -l)
mkdir single_busco_combined
cd single_busco_combined
out_folder=$(pwd)
cd ..
for species_folder in $folder_list
do
species_name=$(echo $species_folder | cut -f1 -d".")
cd $species_folder"/run_arachnida_odb10/busco_sequences"
cp -r single_copy_busco_sequences single_copy_busco_sequences_renamed
cd single_copy_busco_sequences_renamed
file_list=$(ls *.faa)
for i in $file_list;
do
to_be_replaced=$(grep ">" $i)
new_name=">"$species_name
sed -i "s/$to_be_replaced/$new_name/g" $i
done
for i in $file_list
do
cat $i >> $out_folder/$i
done
cd ../../../..
done
Agora podemos aplicar filtros na matriz, por exemplo, removendo genes que tenham menos de 3 terminais, uma vez que não podemos inferir um árvore com eles. Para isso, temos que introduzir uma nova ferramenta: o if statements. Simplificadamente falando, if funciona da seguinte forma: ‘if [ condição ]; then comandos fi’. Para a condição usamos sinas como por exemplo:
-lt menor que (less than)
-gt maior (greater than)
-eq igual (equal)
… dentre outros (veja mais detalhes na Figura 2 e neste link.

Exemplo:
A=100
B=50
if [[ $A -gt $B ]]
then
echo $A é maior que $B
fi
O script abaixo deve ser executado depois (sem fechar o terminal) de ter executado o script que juntarmos todas as espécies em um arquivo por gene, pois utiliza as mesmas variáveis.
cd $out_folder
file_list=$(ls *.*)
for i in $file_list
do
n_seq=$(grep -c ">" $i)
if [[ $n_seq -lt 4 ]]; then
rm $i
fi
done
cd ..
Agora podemos selecionar matrizes de completudes diferentes, por exemplo, excluindo todos os genes que tenham menos de 50% das espécies presentes ou uma matriz 100% completa. (No caso do exemplo, a matriz com 50% não vai existir pois eliminamos tudo que tem menos de 3, mas o script é útil para outros dados.)
cp -r $out_folder $out_folder"_50p"
cd $out_folder"_50p"
file_list=$(ls *.*)
m50p=$(( $n_species / 2 ))
for i in $file_list
do
n_seq=$(grep -c ">" $i)
if [[ $n_seq -lt $m50p ]]; then
rm $i
fi
done
cd ..
cp -r $out_folder $out_folder"_100p"
cd $out_folder"_100p"
file_list=$(ls *.*)
for i in $file_list
do
n_seq=$(grep -c ">" $i)
if [[ $n_seq -lt $n_species ]]; then
rm $i
fi
done
cd ..
Quantos genes temos em cada matriz? Use seus conhecimentos de bash para contar.
Os genes nessas pastas estão prontos para alinhamento. Note que temos matrizes te aminoácidos, não nucleotídeos. Para divergências recentes isso pode não ser muito interessante. Com scripts mais complexos, é possível obter as sequencias de nucleotídeos correspondentes a cada gene.
Obs: Existe um script em python disponibilizado por Jamie McGowan - 2020 neste link para transformar o output do BUSCO para filogenômica.
Alinhamento
Para alinhar podemos usar o mafft em loop nos arquivos das pastas obtidas anteriormente. Para alinhar os genes resultantes do OrthoFinder, execute o loop abaixo na pasta de output do programa
cd ~/filogenomica/data/OrthoFinderResults
cd Results_Dec03
input_folder=Single_Copy_Orthologue_Sequences
outputfolder=$(echo $input_folder"_mafft")
mkdir $outputfolder
cd $input_folder
files=$(ls)
for file in $files
do
echo "Aligning" $file
outname=$(echo $file"-aligned")
mafft --thread 16 --adjustdirectionaccurately --localpair --maxiterate 1000 $file > $outname
mv $outname ../$outputfolder/$file
done
# como utilizamos a função –adjustdirectionaccurately
# temos que remover o _R_ que é inserido quando a sequencia é o complemento reverso.
cd ../$outputfolder
sed -i 's/>_R_/>/g' *.fa
echo "Alignment Finished"
Alinhe as sequencias resultantes do BUSCO. Faça os ajustes necessários nos comandos